宏基因组学(Metagenomics)
- 物种分类数据文件准备
- Windows下16S rDNA扩增子测序分析流程
- While the alpha diversity analysis acts like a summary statistics of a single population, the beta diversity measure acts like a similarity score between populations.
宏基因组数据分析
基于全基因组测序(WGS)的宏基因组学(Metagenomics)是一种以环境样品中的全部微生物的基因组DNA为研究对象,以微生物种群结构、功能基因及其与环境因子相互作用关系等为研究目的的微生物研究方法。WGS宏基因组的分析流程如图1所示:
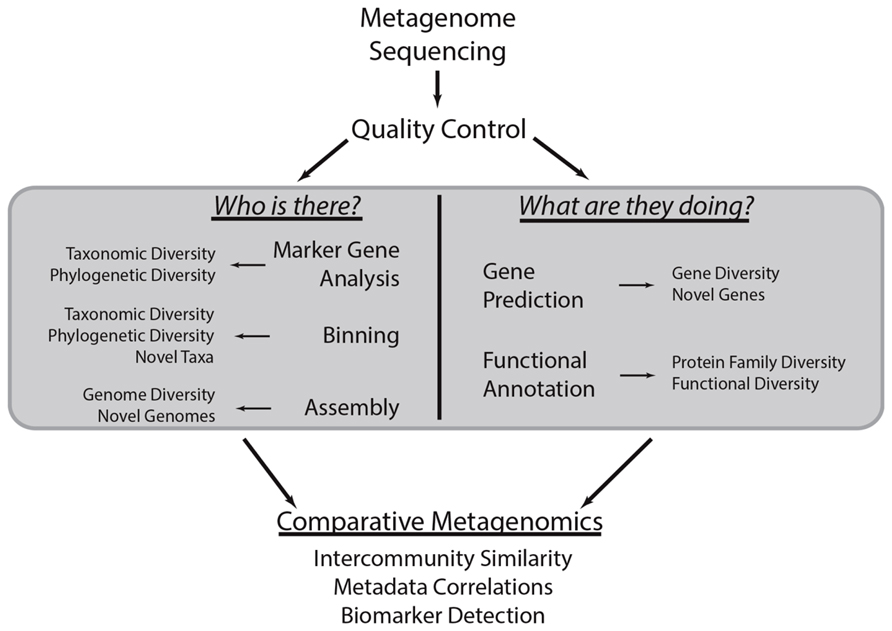
一个典型的宏基因组研究包括5步[2]:
- 样品收集、处理与测序(Experimental pipeline):根据研究内容,从环境样品中直接提取全部DNA,用酶切或超声波打断DNA,构建测序文库,然后上机测序;
- 预处理(Pre-processing):对测序reads进行预处理以去除低质量和污染的序列;
- 分类学、功能、基因序列分析(Sequence analysis):利用组装软件对质控后的序列进行组装,得到contig和scaffold拼接序列,后对scaffold进行基因预测,并通过比对分析和数据库搜索对预测基因进行物种分类和功能注释;
- 后续处理,各种统计分析(Post-processing);
- 实验验证(Validation)。

宏基因组组装
虽然宏基因组学的最终目标是能组装或拼接出一个微生物群落中的所有微生物基因组,但目前受测序技术的限制还远不能达到这个目标。宏基因组测序样本中的微生物数量未知,它们的相对丰度也差异很大,导致其测序的深度也是各不相同。特别是来自复杂微生物群落的样本,其中大多数物种都是低深度的测序,使其组装基本不可能完成。另外相近物种之间的序列可能比较相似,经常导致不同OTUs的序列形成嵌合(chimeric)组装。尽管有这些挑战,宏基因组reads组装是非常重要的分析步骤,特别对复杂度比较低的样品。通过组装能发现未知的微生物基因组(如从Yellowstone lake发现3个新的病毒),或能从新组装的基因组中预测新的基因或功能元件(如细菌的视紫红质(rhodopsin),基因编辑CRISPR片段等)。 目前已开发一些针对宏基因组的专用组装软件,如用于Illumina测序的短reads的组装软件MetaVelvet, metaSPAdes等。这些软件与单基因组组装软件类似都是采用de Bruijn graph算法,具体组装原理可以参见本书第15章相关内容。组装结果经常得到许多小的contigs, 可通过MetaQUAST来评价宏基因组组装质量,像N50及一些汇总统计值,如contigs总数,contig长度的最大,最小与中位值等。检测嵌合组装是质量评价的重要内容,目前还没有合适的工具,但可以通过一些嵌合信号,如覆盖度、GC含量,密码子偏好的变化等来检测嵌合组装。使用paired-end reads与较高的序列相似度域值可以减少嵌合组装。Contig组装后,可以通过将paired-end reads比对回contigs,利用reads之间的连接关系和插入片段大小信息,将contigs拼接成scaffolds,如软件Bambus2可以用各种测序平台的reads来做scaffolding。
微生物群落结构分析
宏基因组研究中最关键的一个目标是揭示样本中微生物群落的结构,并根据物种(或其它分类单元)丰度对其进行量化。理论上宏基因组测序可以获得样本中所有基因组的信息,其中自然包括16S rRNA基因等标记基因的片段。 从测序数据获得物种分类或功能信息有两种方法:短读段直接定位(mapping)与先从头(de novo)组装。这两个方法都依赖序列同源搜索,以reads或组装的contigs在各种基因数据库中搜索序列信息。这个分析过程受已知基因序列信息的限制,但随着测序微生物基因组的快速增加将逐渐缓解。
(1)基于标记基因的分类分析
系统发育标记基因(Phylogenetic gene marker)是指广泛存在于微生物中,序列非常保守,但又有部分可变区域的基因,可以用于分析微生物群落结构与组成。常用的标记基因有核糖体RNA基因(如16S/18S rRNA),或普遍存在于微生物基因组中的单拷贝蛋白质基因(多拷贝基因易混淆丰度估计),如recA (DNA recombinase A), rpoB (RNA polymerase beta subunit), fusA (protein chain elogation factor) 和 gyrB (DNA gyrase subunit B)等。该方法将宏基因组测序读段(reads)与相对较小的标记基因参考数据库进行比对,因此是一种从宏基因组数据快速估计物种多样性的方法。应用标记基因推断一个群落的分类组成有两种通用的方法:第一个是基于序列相似性方法,如MetaPlnAn把宏基因组序列跟一个含系统发育树的特异类群(clade-specific)基因数据库进行同源搜索进行分类注释。MEGAN通过运用最近公共祖先(lowest common ancesters, LCA)算法分析读段与参考基因组的BLAST比对结果进行系统分类。另一个是基于分类标志基因中的系统演化信息方法,通过多序列比对来从宏基因组reads中推断进化关系。如Amphora首先用隐马尔科夫模型(HMMs)来比对宏基因组reads到多个标志基因序列,然后从这个多重比对中构建进化树。PhylOTU通过构建进化树把16S rRNA基因的非重叠同源读段联系起来,随后依据进化距离把这些读段聚类为分类群。
(2)物种分类与分箱
物种分类(taxonomic classification)或分箱(binning)指把宏基因组序列(reads或contigs)依据某些序列特征,利用机器学习方法进行亲缘关系推断的归组过程。不同的分箱(bin)代表序列来至不同的分类群(如属或科等)。分箱后可以了解样本中可能含有的微生物的种类与及其丰度,还能识别未能发现的新物种。分箱的准确性依赖于序列长度,长reads或contigs聚类可以得到较高分辨率的分类,可归类到科(family)或属(genus)水平,而短reads由于信息较少只能归类到门(phylum)水平。 目前分箱方法主要分为两大类,一是依靠分类学的方法,可进一步分为基于序列组成(composition)与基于序列相似性的方法;二是不依赖分类学的方法。
- 基于序列组成的聚类,通过一些序列特征,如GC含量、寡核苷酸序列(k-mers)频率、与密码子偏好等把序列指定到不同的分类单元。这个方法假设来源于相近物种的序列特征也会比较相近。目前这个方法的工具有PhyloPythia, Phymm等。不像其它两种方法,这个方法不用对reads与参考序列进行比较(有些工具也需要用不同分类的参考序列来训练算法)。例如PhyloPythia用不同长度k-mers的出现频率把宏基因组序列指派到不同的分类单元(clades),它是基于支持向量机(SVM)训练已注释的分类物种参考序列的算法。虽然基于序列组成的方法速度比较快,但由于同一物种内或不同物种间的序列组成的差异较大而造成分类的准确度不高。
- 同源聚类法是基于序列相似性,并假设来自相近物种的序列也更相似。这个方法是通过把宏基因组reads序列搜索已经分类注释的微生物数据库来确定它们的分类来源。有许多分类数据库可用,如NCBI RefSeq、Ensembl和UniProt等。如MEGAN将宏基因组序列与NCBI已知分类来源的序列数据库进行BLAST分析。由于需要大量的BLAST搜索,它的计算资源要求比较高,也可以用更高效的短序列比对工具如BWA或bowtie2等代替BLAST。另外,由于是基于已经注释的分类序列,这个方法也不适合发现目前未知的物种或分类(taxa)。针对这个问题,MG-RAST提供了基于最近公共祖先(LCA)算法,使用LCA算法可将未知物种序列分配到最近的公共祖先分类群。由于这个方法也是通过指派宏基因组序列到已知参考基因组的物种,不适合用于研究有许多未知物种的微生物群落。
- 第三种方法是不依赖分类学的方法,不需要用到参考数据库,直接依靠序列特征进行聚类,主要工具有CompostBin、MetaCluster和AbundanceBin等。这类方法不能直接进行分类注释,但可把序列聚类后按类别进行组装获取一个完整的微生物基因组序列,可用于识别未知的新物种。 分类分析需要考虑多种影响因素,如数据类型、可用的参考数据库和训练数据集等。一般来说,短读段的数据由于信息量不足,不适合采用基于序列组成的方法。基于序列相似性的方法可准确分类,但需要依赖可靠的参考数据库,因此无法对未知物种序列进行分类。如果因为读段较短导致分类效果不好时,可考虑先将reads组装成contigs后再进行分类注释,但要注意组装过程中出现拼接错误的可能性。
(3)微生物结构比较分析
不同生境或条件的宏基因组比较分析可以揭示内在的微生物群落结构及其动态变化。然而宏基因组的统计比较相对RNA-Seq比较分析更困难,因为宏基因组比较分析涉及大量的差异(variability)。这些差异有源自生物因素的,如不同样本中的微生物组成差异很大,也可能源自技术原因,如测序深度不够导致不能充分抽样(undersampling)低丰度的物种。在序列抽样过程中,测序量较少的物种更容易受随机(stochastic)因素的影响。一个样品的测序量受多种因素的影响,包括物种的相对丰度,基因组大小,基因组拷贝数,种内异质性,DNA提取方法等。由于受生物或技术因素的限制,经常会出现一些物种(或OTUs)在一个样本中能被检测到,而在另一样本中就没有被检出。如果需要研究稀有物种,可以通过细胞富集技术(如flow cell sorting)人工增加它们的丰度。 与RNA-Seq数据分析类似,宏基因组比较分析前也需要进行数据标准化处理。目前宏基因组还没有统一的标准化方法,一种方法是total-sum scaling(TSS),把指派到某个OTUs的原始reads数量除以样本的reads总数,等同于RNA-Seq的总计数方法。另一个方法是cumulative-sum scaling(CSS),与RNA-Seq中的upper quartile方法类似,把指派到某个OTUs的原始reads数量除以达到某个百分比的累积总数。目前的研究表明CSS比其它标准化方法更有效。 目前有许多工具可以用来比较不同生境或条件的微生物的丰度,包括metagenomeSeq, LEfSe,MEGAN和MG-RAST等。除了用于微生物群落的相对量比较,MEGAN和MG-RAST也可以进行GENE ONTOLOGY(GO)和KEGG途径比较分析不同条件的功能概况。
微生物群落功能注释
宏基因组的功能注释最主要的任务是在大量序列中找出编码蛋白质的序列,并确定其功能。简单来说就是基因预测(gene prediction)与基因注释(gene annotation)两个过程,两者也可合称为功能注释(function annotation)。再下游的代谢通路、比较功能分析等都是在功能注释基础上进行的。
(1)基因预测
从宏基因组组装后得到的contigs或直接从测序reads中鉴定基因或其它基因组元件(如非编码RNA、CRISPR等),是研究一个微生物群落中的物种分类组成和功能的基本步骤。同全基因组测序一样,宏基因组数据也可通过基因预测工具来鉴定基因。很多传统的基因预测工具对序列的长度有要求,因此不适用于短序列。由于宏基因组序列包含的ORFs通常不是全长ORFs,一般传统预测软件也不会对ORFs不完全做罚分。现已有很多适用于宏基因组基因预测的软件,如FragGeneScan(FGS),MetaGeneMark和MetaGeneAnnotator(MGA)等。而预测其它基因组元素(如ncRNA或CRISPR等),也都有相应的工具,如tRNA-SE或CRISPRFinder等。
(2)基因功能注释
对预测的基因做功能注释可以分析微生物群落的功能。基因功能分析可以揭示一个生境的所有基因(或蛋白质),及其参与的各种生命功能,如代谢、信号传导、环境胁迫或毒力等。一般做基因功能注释的策略是把从ORF序列翻译成蛋白质序列,再搜索参考蛋白质数据库或蛋白质家族数据库。蛋白质数据库常用的有COG(Cluster of Orthologous Group), eggNOG,GO (Gene Ontology)和UniRef等,而蛋白质家族数据库有Pfam, FIGfams或TIGRFAMs。软件HMMer可用于在这类数据库中搜索预测蛋白质的同源物,判断蛋白质是否含有某种结构域,进行宏基因组序列的功能注释。对所有宏基因组的基因做数据库搜索是比较耗时的计算过程,可以用网络资源,如MG-RAST, SEED系统或IMG/M等。 另外,可以把预测的蛋白序列搜索代谢数据库(KEGG Orthology或MetaCyc)来做代谢途径的分析,获得微生物群落的代谢通路及其丰度信息。HUMAnN是一个做代谢途径分析的工具,利用BLASTX把宏基因组reads搜索KEGG Orthology数据库获得一个同源蛋白家族的相对丰度,随后采用MinPath把蛋白质家族分配到代谢途径。MinPath是最大简约法来推断所观察到的蛋白家族和它们的丰度,可获得代谢途径的最小子集。经过进一步的降噪处理,HUMAnN的输出可以显示途径的覆盖度(如一个途径是否存在或缺失),和每个途径在样本中的相对丰度(通路的平均拷贝数)。
(3)功能比较分析
由于细菌和古细菌这类微生物编码基因密度较大,平均基因长度较长,因此,宏基因组数据中大多数序列包含编码序列。所以在一定程度上可以忽略物种的影响,直接将微生物群落看作一个整体进行功能比较分析。将基因或基因片段比对至参考数据库,进行标准化后可以计算该基因的相对丰度。这种方法主要用于比较不同样本间的功能家族的相对丰度之间的差异,从而分析其功能差异。 相对丰度的计算是功能比较分析的基础,要注意序列组装可能对频率估计造成影响。因此,这种方法可用未组装的序列来进行,或者是把组装contigs的深度纳入计算。依据参考数据库的不同可以得到不同的丰度谱,例如,比对COGs,Pfam,KEGG,SEED等都可以产生不同类别的丰度谱用于比较。直接比较丰度差异后的,还应作统计学检验以确定这种差异是否具有统计学意义。比较分析结果常用热图(heatmap)直观的可视化展示。此外,主成分分析(PCA)和多维度分析(MDS)等统计方法还可用来分析影响因素。目前有几个常用的软件用于功能比较分析,如Metastats和R软件包ShotgunFunctionalizeR等。
References
- Sharpton Thomas, An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci., 16 June 2014. https://doi.org/10.3389/fpls.2014.00209
- Quince, C., Walker, A., Simpson, J. et al. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35, 833–844 (2017). https://doi.org/10.1038/nbt.3935
- Xinkun Wang, Next-generation sequencing data analysis, 2016, Oxford university press
- Edgar, R.C., 2018. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 34, 2371–2375. https://doi.org/10.1093/bioinformatics/bty113